

概述
本教程将首先建立用于反应力场模拟的Cu离子的水溶液体系,接下来将对文献(van Duin et al., J. Phys. Chem. A, Vol. 114, 9507, 2010)中描述的Cu/O径向分布函数进行计算。
体系构建
首先在一个新的project中建立新模型(CTRL+N)。将该project重新命名为reaxff_models,模型重命名为water。接下来绘制1个水分子。在文件管理器中选中water的模型文件(相应窗口将被激活)。在Sketch toolbar中的下拉列表中选择O(氧),激活位于Toolbar中的Sketch atom工具并在模型窗口中绘制1个O原子。按ESC键退出Sketch atom工具。点击位于工具栏中的Adjust hydrogens按钮添加氢原子。水分子的绘制完成。
为使所构建的模型能够被用于Amorphous Builder,需要为体系指定一个合适的力场(例如Dreiding力场)。在Project manager中选中该模型文件,并点击toolbar中的Assign force field图标。Assign force field对话框弹出。在Force field下拉菜单中选择Dreiding力场,并将Charge calculation前勾选的√去掉。点击OK关闭该对话框。
接下来为该体系构建初始构象。这一步骤将利用Amorphous builder来完成,在Build|Amorphous builder下打开Amorphous builder。在弹出表格中的第一行中选择water模型文件,并将分子数量设定为216个。将Density改为1.002 g/cm 3、Temperature改为300 K,其他默认设置不变。点击Build。几秒钟后会在Project manager中生成1个带有1个新模型文件的project,新project名为Amorphous project,新的模型文件名为Amorphous model。将其分别更名为reaxff和CuH2O。
打开CuH2O模型文件的窗口查看构象。Cu原子的添加方法为从Sketching toolbar栏(可参见sketching)的构建下拉列表中选中Cu,激活位于Toolbar中的Sketching atom图标后在该模型窗口中绘制一个铜原子。完成上述步骤后按ESC键退出Sketch atom工具。
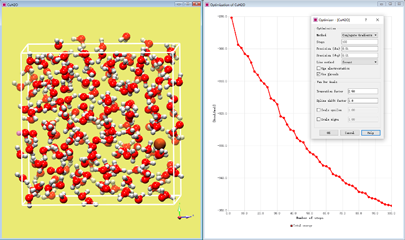
为消除可能的原子重叠,需要对体系进行结构优化。将力场设定为UFF力场,去掉Charge calculation前勾选的√。打开MAPS的结构优化对话框(在Project manager中选中模型文件,单击鼠标右键在出现的快捷菜单中选择Optimizer)。方法选择Conjugate Gradients,设定步数为100后勾选Use threads。点击OK。优化将持续几秒钟。接下来的步骤将用到该优化构象。
LAMMPS的计算设置
选中Project manager中的CuH2O模型文件,然后右键点击,Simulate with→LAMMPS。屏幕中将弹出LAMMPS对话框。在Types of system中选择Reactive并在Ensemble中选择NVT。点击Next继续至下一步。
随后将出现Reactive的对话框,在这里可为反应力场的模拟设置基本输入。点击Selective按钮然后导航至含有ffield.reax.CuOH2O的路径下并选取。该文件中含有van Duin等人在文献中使用的参数设置。在页面底端的表格中可以查看参数文件所支持的元素对模拟盒子中原子类型的定义。点击Next继续至下一步。
在弹出的Molecular dynamics对话框中将Duration of run设为120 ps、Time step设为0.5 fs。点击Next下一步。
在弹出的Output对话框中将Write properties every和Write trajectory every修改为1000步(即50 fs,240个构象)。
点击Next。在弹出的Reative output对话框中将Write bonds file every改为1000步。点击Finish按钮开始运行计算。当弹出Start LAMMPS now?的提示时选择Yes。
注意:
在用ReaxFF进行模拟时,由于每一步都会对电荷与键级进行计算,因此应将时间间隔尽可能设短。根据van Duin等人在文献中的报道,为保证相空间中有效覆盖到的碰撞和反应的连续性(efficient coverage of the phase space allowing collisions and reactions to occur smoothly),模拟的时间步长应设为0.1 fs。
结果分析
模拟过程需要几小时。在MAPS上加载模拟的输出文件并查看轨迹文件。由于体系中不存在化学键,用球棍模式展示并将原子置于盒子中分析效果更佳。在输出的模型文件上单击鼠标右键,并依次选择Analyse with→Trajectory analysis打开对话框。在Structure选项卡中勾选Radial distribution function前的方框。将最大距离设置为5.0Å,步长设置为0.1Å后点击Apply。在生成的图中用右键快捷菜单选择Cu-O对。输出的曲线应如下图所示。
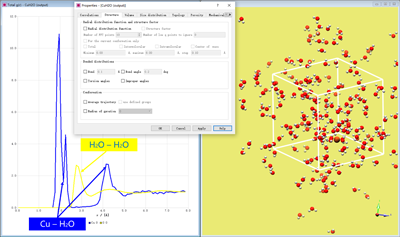
Cu-O径向分布函数
RDF清楚的展示出2个水化层。第1个水化层有2个峰分别位于1.94 Å和2.25Å处,分别对应Cu-Oeq和Cu-Oax。第2个水化层有1个峰位于4.20Å处。上述结果与van Duin等人报道的结果是一致的。说明模拟使用的势能能够很好的反映铜水合物的Jahn-Teller效应,这是由于水分子的特点是更倾向于采取横向位(eq)或纵向位(ax),而非介于两者之间。
MAPS材料及化工过程设计平台是一款多尺度、可扩展的平台;可应用于从量子化学计算到中尺度计算。MAPS适合描述含能材料、离子液体、高分子材料、合金材料、复合材料、电池材料等性质。
MAPS-ReaxFF:反应动力学分析模块擅长研究多相催化、含能材料、电池电极材料、汽车尾气净化、沥青、高分子及煤的性能。理论上描述催化反应路径、高温裂解和复杂合金中载流子扩散性能等。
Amorphous Builder无定形结构建模模块:最大的特点是建模效率高,应用于高分子、流体、纳米结构、石油、橡胶、气固液混合结构、离子液体、电解质和表面活性剂等模型。
· 支持丰富的模型文件格式
· 自动识别原子类型,方便赋予分子力场
· 图形界面帮助下生成 data 和 in 输入文件
· 动画演示动力学变化过程,并输出为可用于 ppt 的演示的 swf、mpg、mp4 和 wmv 格式的文件。
· 根据动力学轨迹分析多种动力学过程描述性质,偶极矩分析、相关函数分析、均方根 计算、扩散系数分析、径向分布函数分析、键长等性质分布分析、回转半径分析和自由体积预 测等。径向分布函数包括分子间分子内(原子间)径向分布函数。
· 粘度计算:通过计算应力自相关函数预测材料的粘度。
· 密度梯度分析
· 预测玻璃化转变温度(Tg)
MAPS软件
云榜(北京)科技有限公司
· 地址:北京市海淀区永澄北路2号院1号楼A座五层530室
· 邮箱:sales@scisim.com.cn
· 公司座机:010-53387159


扫一扫
查看手机端官网
