用于研究化学反应、结构、电子和其他分子性质的从头算软件。
特性&功能
Hartree-Fock(HF),密度泛函理论(DFT),时变DFT,Møller-Plesset微扰理论(MP2)和耦合聚类计算(CCSD和CCSD(T))。不同近似水平下的总能量,电荷密度,结构,电子,光学和热力学性质。
概述
NWChem提供分子性质的精确计算:电子结构、键长、键角、电学、振动、光学(IR、RAMAN、UV-Vis光谱)和其他性质,以及沿着反应路径的能量学(过渡态和能垒)。NWChem允许科学家在不同的理论水平上工作,包括HF、DFT、TD-DFT、MP2、CCSD和CCSD(T),并使用大量的基组和有效的核心电位。支持分子动力学计算,可以模拟动力学现象和构象搜索的性能。
NWChem非常适合解决气相化学问题,在均相催化等领域探索化学反应性。此外,NWChem能够通过COSMO模型模拟溶剂效应。
NWChem提供了使用RPA和Tamm-Danconff近似研究激发态(单重态和三重态)以及计算CD谱的能力。最后,NWChem可以通过TRANSITION STATE LOCATOR,使用线性/二次同步渡越(LST/QST)或微动弹性带(NEB)来定位过渡态。
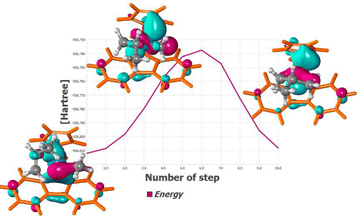
用NWChem研究的Ziegler-Natta均相催化形成间规聚丙烯。
参考文献
[1] M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus, W. A. de Jong, “NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations” Comput. Phys. Commun. 181, 1477 (2010)



