利用经典分子动力学(MD)模拟研究大规模反应体系,延长QM模型的观察时间。
特性&功能
分析LAMMPS ATOMISTIC/REAXFF模拟燃烧、氧化和聚合等反应的结果,以研究各种参数(如温度、压力、成分)的影响,并获得一个反应如何在更大范围内发生的信息。
概述
Reactive Force Field(ReaxFF)是一个依赖于键序的力场,在分子动力学模拟中允许键的产生和解离。ReaxFF集成在LAMMPS模拟引擎中,支持仅由几个原子到数百万个原子组成的反应体系建模。LAMMPS插件提供了对ReaxFF参数化的直接访问。ReaxFF模拟根据每个时间步长的键和键序生成原始连接数据,但没有直接参考分子种类。
REAXFF ANALYSIS对系统进行重构,以追踪模拟过程中产生或破坏的分子种类。用户以表格形式查看结果并选择感兴趣的种类,以便可视化它们的三维结构,以及查看其种类在模拟过程中如何演变的图像。
与此同时,用户可以访问所选种类的反应途径,了解中间步骤,估计完成反应所需的时间。支持下列反应:
● 冲击效应研究
● 碳氢化合物氧化
● 碳氢化合物热分解
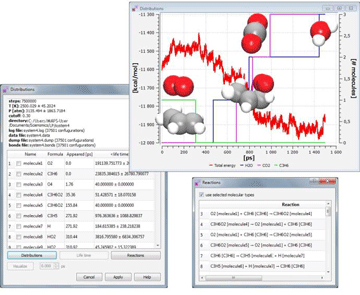
丁烯氧化,1.5 ns后得到特征构
参考文献
[1] C. T. van Duin, S. Dasgupta, F. Lorant, W. A. Goddard III, 2001. J. Phys. Chem. A, 105, pp. 9396-9409.
[2] T. R. Mattsson et al., 2010. Phys. Rev. B, 81, 054103.
[3] J. Budzien, A. P. Thompson, S. V. Zybin, 2009. J. Phys. Chem., 113, p. 13142.
[4] L. Zhang et al., 2009. J. Phys. Chem. A, 113, pp.10619-10640.
[5] K. Chenoweth, A. C. T. van Duin, W. A. Goddard, 2008. J. Phys. Chem. A, 112, pp. 1040-1053.
[6] M. R. Weismiller, A. C. T. van Duin, J. Lee, R. A. Yetter, 2010. J. Phys. Chem. A, 114 (17), pp. 5485-5492.
[7] J. A. Keith, D. Fantauzzi, T. Jacob, A. C. T. van Duin, 2010. Phys Rev B, 81, p. 235404.
[8] M. Aryanpour, A. C. T. van Duin, J. D. Kubicki, 2010. Phys. Chem. A, 114 (21), pp. 6298-6307.
[9] K. Chenoweth et al., 2008. J. Phys. Chem. C, 112 (37), pp. 14645-14654.
[10] D. Raymanda et al., 2008. Surface Science, 602, p. 1020.



