

概述
工业应用
石油天然气及石油化工、精细化工品、汽车及运输
挑战
考察Ni、Pt的单一或双金属对氨的催化解离
模拟内容
设计Ni、Pt的单一或双金属表面
计算氮原子的结合能
模拟结果
基于密度泛函理论计算了氮原子在单一或双组分催化剂上的吸附。Ni-Pt-Pt单层双金属表面被认为对于氨气解离具有最高催化活性,该认识与实验结果相一致。
类型
工业应用
软件
Quantum
QUANTUM ESPRESSO
Engineering
MAPS
性质
吸附热
应用
催化剂
金属

近年来通过氨气解离将氨气作为储氢媒介成为极具吸引力的研究方向。该反应第一步为氨气脱氢,随后为N与H重组生成相应的N2与H2。氮原子与表面的结合能,一方面应足够强,以利于进行氨气的解离;另一方面又应足够弱,以利于氮原子重新结合,并从表面脱离,从而完成整个催化过程。该过程的氮结合能与氨解离活性存在火山型曲线,其中钌位于该火山曲线的顶点。尽管其被认为作为单金属组分具有最高的解离活性,然而价格昂贵且产量有限。因此通过开发非贵金属或金属合金进行催化剂改性极为重要。
挑战
单层双组分催化剂是一种特定类型的双金属催化剂,在主体的顶层中分布有第二种金属。增加的层级可位于主体金属的表层,增加了表面构象,也可位于表层下方,形成次表层构象。此类体系的性质与改性金属及主体金属自身的性质与所处的位置有关,与相应主体金属或合金体系的性质存在着巨大差异。由于该非线性特性的存在,此类贵金属催化剂尚无常规的设计方法可实现。
模拟工作
在该研究案例中,我们基于密度泛函理论(DFT),利用MAPS的Quantum Espresso模块对氮原子在4种不同单一及双组分金属催化剂上的吸附进行了考察:Ni(111)、Pt(111)、Ni-Pt-Pt 及Pt-Ni-Pt表面。自旋极化DFT计算选用PBE 交换关联方程及PAW赝势,能量截断设置为600 eV、Brillouin 采样的k点设置为3 ´ 3 ´ 1。费米拖尾设置为0.2 eV。Ni(1 1 1)及Pt(1 1 1)的表面通过对相应晶胞切割后扩胞为3 ´ 3 ´ 1超胞,利用4层金属片层并沿z方向构建15 Å真空。

Figure 1:单一或双金属催化剂的结构优化:a) Ni(111)表面;
b) Ni-Pt-Pt表面;c) Pt-Ni-Pt表面;d) Pt(111)表面。
模拟结果
Ni-Pt-Pt及Pt-Ni-Pt的单层双金属催化剂表面结构优化结果如Figure 1所示。N原子吸附与优化后表面的hollow位,经计算吸附能Eads = E(surface+N) – E(surface) – E(N)。上述4个体系的吸附能及氮-金属间距离列于Table 1。
吸附体系 | N 吸附能(kcal mol-1) | d(N,M) (Å) | ||
| 本研究 | 前人研究 | 本研究 | 前人研究 |
Pt | -102.8 | -102.1 (0.7%) | 1.97 | 1.95 (0.7%) |
Ni | -123.4 | -113.8 (8.4%) | 1.77 | 1.77 (0.1%) |
Ni-Pt-Pt | -136.6 | -130.7 (4.5%) | 1.77 | 1.76 (0.1%) |
Pt-Ni-Pt | -90.5 | -87.5 (3.4%) | 1.98 | 1.94 (2.0%) |
本研究计算得到的吸附能及氮-金属距离数值与前人的结果相一致。Ni-Pt-Pt催化剂被认为具有最高活性,吸附能接近单金属Ru的最佳吸附能数值~134 kcal mol-1。该结论与实验测得的Ni-Pt-Pt双金属单层表面比单金属Ru催化剂具有更高活性相一致。Figure 2展示了N在Ni-Pt-Pt表面吸附的稳定构象。
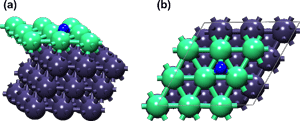
Figure 2:N在Ni-Pt-Pt表面吸附优化构象的侧视图(a)及俯视图(b)
对单一及双金属表面的计算研究表明在展开实验设计前,可利用氮结合能作为描述符对氨气解离理想催化剂活性加以考察。
Hansgen, D. A.; Vlachos, D. G.; Chen, J. G. Nat. Chem. 2010, 2 (6), 484–489. https://doi.org/10.1038/nchem.626.
建模功能:晶体模型、无定型结构模型、表面和界面模型、聚合物模型、交联模型及特殊模型;
分析功能:自动从结果提取关键性质、粘度分析、密度分布分析、孔径分析、径向分布函数、均方根位移分析、扩散系数分析、相关函数分析、反应产物片段分析、基元反应分析。
MAPS-Quantum Espresso:借助 MAPS 强大的平台功能,基于图形界面的方便工具直接生成输入文件,并对结果进行分析。
Quantum Espresso是一款强大的平面波代码,用于在纳米尺度模拟的性质。使用密度泛函理论(LDA, GGA, meta-GGA)和赝势方法(超软赝势、PAW、模守恒赝势)。基于不同密度泛函方法,可进行电子结构、分子动力学、微扰计算,研究材料的化学反应、动力学、弛豫、电场线性响应、离子位移、光学性质或磁性、超声转变温度、电声耦合等。
MAPS软件
云榜(北京)科技有限公司
· 地址:北京市海淀区永澄北路2号院1号楼A座五层530室
· 邮箱:sales@scisim.com.cn
· 公司座机:010-53387159


扫一扫
查看手机端官网
