MAPS — 最新材料设计平台发布!
来源:
|
作者:pmo878164
|
发布时间: 2016-09-08
|
1188 次浏览
|
分享到:
MAPS(Materials And Processes Simulations)是集中多款优秀的第三方材料设计软件的具有很强扩展性的材料设计平台。
MAPS 概述
Materials And Processes Simulation
MAPS(Materials And Processes Simulations)是集中多款优秀的第三方材料设计软件的具有很强扩展性的材料设计平台。
MAPS可应用于从量子化学计算到中尺度计算,在不同时间和空间尺度上真正的实现了多尺度计算平台建设的目标。MAPS包括友好的图形用户界面供用户建模、方便分析结算结果数据,并整合了大学和研究所开发的被广大世界科研用户肯定的计算模拟软件。
MAPS是一种科研平台,可以计算高分子的物性,小分子和高分子凝聚状态和变化分析,研究分子反应机理,固体、表面、界面的电子结构计算。
MAPS中各种计算引擎由开发源程序和与其相对应的界面组成的Plug-in组成。可以通过在基本MAPS模块中添加不同Plug-in来实现研究不同体系和时间尺度的目的。
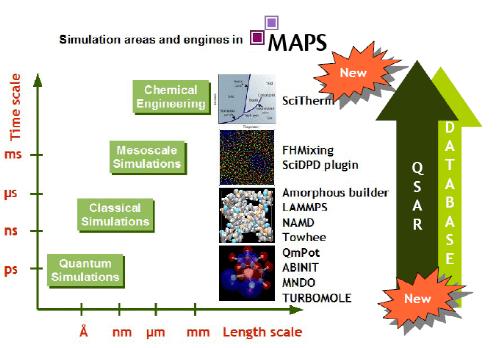
MAPS中的计算引擎
* ABINIT,是由开源代码项目负责开发的第一原理能带计算软件。它包含众多密度泛函方法中最新的技术,包括TDFT、GW、PAW等。研究金属、陶瓷、半导体等的结构、电子状态、物性等方面有独到的优势。
* Amorphous Builder, 是由雅典工大的Doros Theodorou教授与Scienomics公司共同合作开发的无定形结构建模软件。采用了CBMC(Configurational Bias Monte Carlo)建模算法,可以非常高效的得到合适的最终构型。
* FHMixing, FHMixing是基于Molecular Silverware法的二元混合物片段间相互作用计算软件。用于高分子、液体的热力学物性和SciDPD的相互作用参数预测。
* LAMMPS-Atomistic,是由美国能源部和企业组成的CRADA(Cooperative Research and Development Agreement)开发的分子动力学计算程序。支持各种各样的边界条件,并能处理包括分子、高分子、生物分子、粉末等广泛的体系研究。
* MNDO, 是由马克思-普朗克煤炭研究所得Walter Thiel教授研究组开发的半经验分子轨道计算软件。能预测由小分子到比较大的分子的结构和物性。
* NAMD,NAMD是由美国伊利诺伊大学理论生物物理研究做开发的分子动力学软件。主要应用于生物分子、合成高分子等的分子动
力学计算。
* QMPOT,QmPotSauer教授研究组开发的QM/MM计算软件。可以进行组合TURBOMOLE、MNDO、ABINIT、LAMMPS的多尺度计
算。QmPot不仅能做QM/MM计算,而且能与TUBOMOLE和ABINIT组合进行QM/MM计算。
* LAMMPS-DPD,SciDPD是耗散粒子动力学软件。分子动力学难以处理的Meso尺度计算机模拟SciDPD能处理的很好,如液体或者高分子的相分离等。
* SciTherm,是由Scienomics研发中心完全自主开发的一款计算引擎。SciTherm实现了通过统计缔合流体理论状态方程获得最先进
的热力学模型。SciTherm为过程模拟中提供可靠的物理性质预测数据。其应用领域涵盖了多个工业领域,如化工、石油、分离技术、
制药工业等等。
* SciPharma,基于PC-SAFT 计算药物分子溶解度的工具。采用创新性的算法确定药物分子的PC-SAFT 方程中的参数,并基于此状态方程 计算纯态和混合溶剂的溶解度。
* Towhee,是由美国明尼苏达大学J. Lija Siepmen教授的小组开发的蒙特卡洛计算软件。运用计算效率更高的分子构象样本技术,
并支持各种力常数。可方便的处理分子动力学很难处理的液液、气液两相平衡等,及沸石的吸附现象研究等。
* Turbomole, 是由卡尔斯鲁大学理论化学研究组开发的量子化学计算软件,支持HF、MP2、DFT、TDFT、CC等各种方法。特别适合研究分子结构、反应机理预测等.



